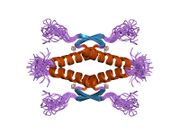
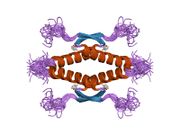
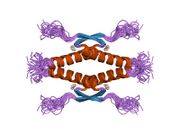
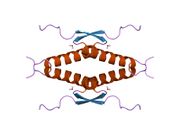
p53
| edit |

p53 (also known as protein 53 or tumor protein 53), is a tumor suppressor protein that in humans is encoded by the TP53 gene.[1][2][3] p53 is important in multicellular organisms, where it regulates the cell cycle and, thus, functions as a tumor suppressor that is involved in preventing cancer. As such, p53 has been described as "the guardian of the genome", the "guardian angel gene", and the "master watchman", referring to its role in conserving stability by preventing genome mutation.[4]
The name p53 is in reference to its apparent molecular mass: It runs as a 53-kilodalton (kDa) protein on SDS-PAGE. But, based on calculations from its amino acid residues, p53's mass is actually only 43.7 kDa. This difference is due to the high number of proline residues in the protein, which slow its migration on SDS-PAGE, thus making it appear heavier than it actually is.[5] This effect is observed with p53 from a variety of species, including humans, rodents, frogs, and fish.
Contents |
Nomenclature
P53 is also known as:
- UniProt name: Cellular tumor antigen p53
- Antigen NY-CO-13
- Phosphoprotein p53
- Transformation-related protein 53 (TRP53)
- Tumor suppressor p53
Gene
In humans, p53 is encoded by the TP53 gene located on the short arm of chromosome 17 (17p13.1).[1][2][3] TP53 orthologs [6] have been identified in most mammals for which complete genome data are available.
In humans, the two most common polymorphisms to occur involve the substitution of an Arginine base for a Proline base. This polymorphism arises out of a SNP mutation on the 72 codon, where a guanine base is replaced by a cytosine (http://www.ncbi.nlm.nih.gov/pubmed/9607760)
For these mammals, the gene is located on different chromosomes:
- Chimp and orangutan, chromosome 17
- Macaque, chromosome 16
- Mouse, chromosome 11
- Rat, chromosome 10
- Dog, chromosome 5
- Cow, chromosome 19
- Pig, chromosome 12
- Horse, chromosome 11
- Opossum, chromosome 2
(Italics are used to denote the TP53 gene name and distinguish it from the protein it encodes.)
Structure
Human p53 is 393 amino acids long and has seven domains:
- N-terminal transcription-activation domain (TAD), also known as activation domain 1 (AD1), which activates transcription factors: residues 1-42.
- activation domain 2 (AD2) important for apoptotic activity: residues 43-63.
- Proline rich domain important for the apoptotic activity of p53: residues 64-92.
- central DNA-binding core domain (DBD). Contains one zinc atom and several arginine amino acids: residues 100-300.
- nuclear localization signaling domain, residues 316-325.
- homo-oligomerisation domain (OD): residues 307-355. Tetramerization is essential for the activity of p53 in vivo.
- C-terminal involved in downregulation of DNA binding of the central domain: residues 356-393.[7]
A tandem of nine-amino-acid transactivation domains (9aaTAD) was identified in the AD1 and AD2 regions of transcription factor p53.[8] KO mutations and position for p53 interaction with TFIID are listed below:[9]
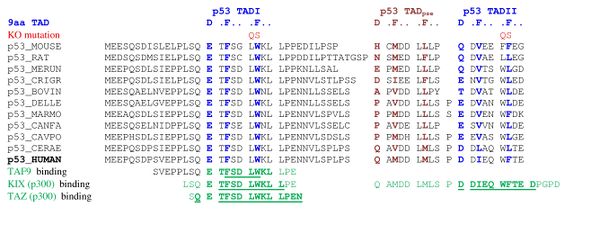
9aaTADs mediate p53 interaction with general coactivators - TAF9, CBP/p300 (all four domains KIX, TAZ1, TAZ2 and IBiD), GCN5 and PC4, regulatory protein MDM2 and replication protein A (RPA).[10][11]

Mutations that deactivate p53 in cancer usually occur in the DBD. Most of these mutations destroy the ability of the protein to bind to its target DNA sequences, and thus prevents transcriptional activation of these genes. As such, mutations in the DBD are recessive loss-of-function mutations. Molecules of p53 with mutations in the OD dimerise with wild-type p53, and prevent them from activating transcription. Therefore OD mutations have a dominant negative effect on the function of p53.
Wild-type p53 is a labile protein, comprising folded and unstructured regions that function in a synergistic manner.[12]
Function
P53 has many anticancer mechanisms, and plays a role in apoptosis, genetic stability, and inhibition of angiogenesis. In its anti-cancer role, p53 works through several mechanisms:
- It can activate DNA repair proteins when DNA has sustained damage.
- It can induce growth arrest by holding the cell cycle at the G1/S regulation point on DNA damage recognition (if it holds the cell here for long enough, the DNA repair proteins will have time to fix the damage and the cell will be allowed to continue the cell cycle).
- It can initiate apoptosis, the programmed cell death, if DNA damage proves to be irreparable.
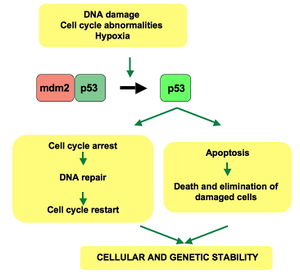
Activated p53 binds DNA and activates expression of several genes including WAF1/CIP1 encoding for p21. p21 (WAF1) binds to the G1-S/CDK (CDK2) and S/CDK complexes (molecules important for the G1/S transition in the cell cycle) inhibiting their activity.
When p21(WAF1) is complexed with CDK2 the cell cannot continue to the next stage of cell division. A mutant p53 will no longer bind DNA in an effective way, and, as a consequence, the p21 protein will not be available to act as the "stop signal" for cell division. Thus, cells will divide uncontrollably, and form tumors.[13]
Recent research has also linked the p53 and RB1 pathways, via p14ARF, raising the possibility that the pathways may regulate each other.[14]
P53 by regulating LIF has been shown to facilitate implantation in the mouse model and possibly in humans.[15]
P53 expression can be stimulated by UV light, which also causes DNA damage. In this case, p53 can initiate events leading to tanning.[16][17]
Regulation
P53 becomes activated in response to myriad stress types, which include but is not limited to DNA damage (induced by either UV, IR, or chemical agents such as hydrogen peroxide), oxidative stress,[18] osmotic shock, ribonucleotide depletion, and deregulated oncogene expression. This activation is marked by two major events. First, the half-life of the p53 protein is increased drastically, leading to a quick accumulation of p53 in stressed cells. Second, a conformational change forces p53 to be activated as a transcription regulator in these cells. The critical event leading to the activation of p53 is the phosphorylation of its N-terminal domain. The N-terminal transcriptional activation domain contains a large number of phosphorylation sites and can be considered as the primary target for protein kinases transducing stress signals.
The protein kinases that are known to target this transcriptional activation domain of p53 can be roughly divided into two groups. A first group of protein kinases belongs to the MAPK family (JNK1-3, ERK1-2, p38 MAPK), which is known to respond to several types of stress, such as membrane damage, oxidative stress, osmotic shock, heat shock, etc. A second group of protein kinases (ATR, ATM, CHK1 and CHK2, DNA-PK, CAK) is implicated in the genome integrity checkpoint, a molecular cascade that detects and responds to several forms of DNA damage caused by genotoxic stress. Oncogenes also stimulate p53 activation, mediated by the protein p14ARF.
In unstressed cells, p53 levels are kept low through a continuous degradation of p53. A protein called Mdm2 (also called HDM2 in humans), which is itself a product of p53, binds to p53, preventing its action and transports it from the nucleus to the cytosol. Also Mdm2 acts as ubiquitin ligase and covalently attaches ubiquitin to p53 and thus marks p53 for degradation by the proteasome. However, ubiquitylation of p53 is reversible. A ubiquitin specific protease, USP7 (or HAUSP), can cleave ubiquitin off p53, thereby protecting it from proteasome-dependent degradation. This is one means by which p53 is stabilized in response to oncogenic insults.
Phosphorylation of the N-terminal end of p53 by the above-mentioned protein kinases disrupts Mdm2-binding. Other proteins, such as Pin1, are then recruited to p53 and induce a conformational change in p53, which prevents Mdm2-binding even more. Phosphorylation also allows for binding of trancriptional coactivators, like p300 or PCAF, which then acetylate the carboxy-terminal end of p53, exposing the DNA binding domain of p53, allowing it to activate or repress specific genes. Deacetylase enzymes, such as Sirt1 and Sirt7, can deacetylate p53, leading to an inhibition of apoptosis.[19] Some oncogenes can also stimulate the transcription of proteins which bind to MDM2 and inhibit its activity.
Role in disease
If the TP53 gene is damaged, tumor suppression is severely reduced. People who inherit only one functional copy of the TP53 gene will most likely develop tumors in early adulthood, a disease known as Li-Fraumeni syndrome. The TP53 gene can also be damaged in cells by mutagens (chemicals, radiation, or viruses), increasing the likelihood that the cell will begin decontrolled division. More than 50 percent of human tumors contain a mutation or deletion of the TP53 gene.[20] Increasing the amount of p53, which may initially seem a good way to treat tumors or prevent them from spreading, is in actuality not a usable method of treatment, since it can cause premature aging.[21] However, restoring endogenous p53 function holds a lot of promise.[22] Loss of p53 creates genomic instability that most often results in the aneuploidy phenotype.[23]
Certain pathogens can also affect the p53 protein that the TP53 gene expresses. One such example, human papillomavirus (HPV), encodes a protein, E6, which binds the p53 protein and inactivates it. This, in synergy with the inactivation of another cell cycle regulator, pRb, by the HPV protein E7, allows for repeated cell division manifested in the clinical disease of warts. Certain HPV types, in particular types 16 and 18, can also lead to progression from a benign wart to low or high-grade cervical dysplasia, which are reversible forms of precancerous lesions. Persistent infection of the cervix over the years can cause irreversible changes leading to carcinoma in situ and eventually invasive cervical cancer. This results from the effects of HPV genes, particularly those encoding E6 and E7, which are the two viral oncoproteins that are preferentially retained and expressed in cervical cancers by integration of the viral DNA into the host genome.[24]
In healthy humans, the p53 protein is continually produced and degraded in the cell. The degradation of the p53 protein is, as mentioned, associated with MDM2 binding. In a negative feedback loop, MDM2 is itself induced by the p53 protein. However, mutant p53 proteins often do not induce MDM2, and are thus able to accumulate at very high concentrations. Worse, mutant p53 protein itself can inhibit normal p53 protein levels.
Discovery
P53 was identified in 1979 by Lionel Crawford, David P. Lane, Arnold Levine, and Lloyd Old, working at Imperial Cancer Research Fund (UK) Princeton University/UMDNJ (Cancer Institute of New Jersey), and Sloan-Kettering Memorial Hospital, respectively. It had been hypothesized to exist before as the target of the SV40 virus, a strain that induced development of tumors. The TP53 gene from the mouse was first cloned by Peter Chumakov of the Russian Academy of Sciences in 1982,[25] and independently in 1983 by Moshe Oren (Weizmann Institute).[26] The human TP53 gene was cloned in 1984.[1]
It was initially presumed to be an oncogene due to the use of mutated cDNA following purification of tumour cell mRNA. Its character as a tumor suppressor gene was finally revealed in 1989 by Bert Vogelstein working at Johns Hopkins School of Medicine.[27]
Warren Maltzman, of the Waksman Institute of Rutgers University first demonstrated that TP53 was responsive to DNA damage in the form of ultraviolet radiation.[28] In a series of publications in 1991-92, Michael Kastan, Johns Hopkins University, reported that TP53 was a critical part of a signal transduction pathway that helped cells respond to DNA damage.[29]
In 1992, Wafik El-Deiry when he was working with Bert Vogelstein at Johns Hopkins University identified the consensus sequence, to which human p53 could bind, by immunoprecipitating human genomic DNA that could be bound by baculovirus-produced human p53 protein. This sequence was published in the first issue of the journal Nature Genetics in 1992 in work that is highly cited. The consensus sequence is 5'-RRRCWWGYYY-N(0-13)-RRRCWWGYYY-3' and is located in the regulatory regions of genes that are activated by the p53 transcription factor. The presence of p53 response elements in or around genes (promoters, upstream sequences, introns) is a powerful predictor of regulation and activation of a particular gene by p53.
In 1993, p53 was voted molecule of the year by Science magazine.[30]
That same year, 1993, Wafik El-Deiry when he was working with Bert Vogelstein at Johns Hopkins University discovered p21(WAF1) as a gene regulated directly by p53. This work was reported in the most highly cited paper ever published in the journal Cell, and provided a molecular mechanism by which mammalian cells undergo growth arrest when damaged. The p21(WAF1) protein binds directly to cyclin-CDK complexes that drive forward the cell cycle and inhibits their kinase activity thereby causing cell cycle arrest to allow repair to take place. p21 can also mediate growth arrest associated with differentiation and a more permanent growth arrest associated with cellular senescence. The p21 gene contains several p53 response elements that mediate direct binding of the p53 protein, resulting in transcriptional activation of the gene encoding the p21(WAF1) protein.
Interactions
P53 has been shown to interact with
- HIPK1[31]
- Replication protein A1[32][33]
- ERCC6[34][35]
- TSG101[36]
- Protein kinase R[37]
- CFLAR[38]
- XPB[34]
- CREB binding protein[39][40][41]
- CREB1[41]
- Mitogen-activated protein kinase 9[42][43]
- Prohibitin[44]
- RPL11[45]
- Ataxia telangiectasia mutated[46][47][48][49][50]
- DNA-PKcs[50][51][52]
- ATF3,[53][54]
- HIPK2[55][56]
- HIF1A[57][58][59][60]
- MED1[61][62]
- Zif268[63]
- GPS2[64]
- EFEMP2[65]
- Promyelocytic leukemia protein[66][67][68]
- PLK3[69][70]
- TP53INP1[71][72]
- Aurora A kinase[73]
- BRE[74]
- PTTG1[75]
- CHEK1[51][76][77] CCNG1,[78]
- Cyclin H[79]
- TOP1[80][81]
- PIAS1[65][82] CDC14B[83]
- BRCA2[74][84]
- BRCA1[74][85][86][87][88]
- RCHY1[89][90]
- CDC14A[83]
- E4F1[91][92]
- PARP1[93][94]
- ZNF148[95]
- HMGB1[96][97]
- Multisynthetase complex auxiliary component p38[98]
- PRKRA[99]
- Ataxia telangiectasia and Rad3 related[47][50]
- TP53BP1[77][100][101][102][103][104][105]
- Cdk1[106][107]
- TP53BP2,[105][108] TOP2B,[109]
- TOP2A[109]
- Bloom syndrome protein[77][110][111][112]
- BAK1[113]
- PARC[114]
- RAD51[74][115][116]
- BARD1[74]
- PSME3[117]
- Aprataxin[93]
- S100B,[118]
- UBE2I[65][119][120][121]
- PIN1[122][123]
- Small ubiquitin-related modifier 1[119][124]
- Huntingtin[125]
- PTEN[126]
- KPNB1[127]
- Cyclin-dependent kinase 7[79][128]
- ING5[129] ELL,[130]
- PTK2[131]
- NUMB[132]
- ING1[133][134]
- P16[45][91][135]
- USP7[136]
- ING4[129][137]
- YWHAZ[138]
- UBE2A[139]
- Ubiquitin C[98][117][124][140][141][142][143][144]
- Werner syndrome ATP-dependent helicase[112][145]
- GNL3[146]
- BRCC3[74]
- CCAAT/enhancer binding protein zeta[147]
- WWOX[148]
- Y box binding protein 1[149][150]
- IκBα[151]
- TATA binding protein[152][153]
- HSPA9[154]
- MDM4[155][156]
- Mdm2[39][45][66][126][133][135][157][158][159][160][161][117][162][132][163][164][165][166][167][168][169][170][171]
- GSK3B[172]
- SMN1[173]
- Heat shock protein 90kDa alpha (cytosolic), member A1[127][174][175]
- TFAP2A[176]
- ANKRD2[149]
- PLAGL1[177]
- Nucleolin[178]
- NDN[179]
- SMARCB1[180]
- EP300[40][159][181][182]
- SMARCA4,[180]
- MNAT1[128]
- TFDP1[183]
References
- ↑ 1.0 1.1 1.2 Matlashewski G, Lamb P, Pim D, Peacock J, Crawford L, Benchimol S (December 1984). "Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene". EMBO J. 3 (13): 3257–62. PMID 6396087.
- ↑ 2.0 2.1 Isobe M, Emanuel BS, Givol D, Oren M, Croce CM (1986). "Localization of gene for human p53 tumour antigen to band 17p13". Nature 320 (6057): 84–5. doi:10.1038/320084a0. PMID 3456488.
- ↑ 3.0 3.1 Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B (June 1991). "Identification of p53 as a sequence-specific DNA-binding protein". Science (journal) 252 (5013): 1708–11. PMID 2047879. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=2047879.
- ↑ Read, A. P.; Strachan, T. (1999). "Chapter 18: Cancer Genetics". Human molecular genetics 2. New York: Wiley. ISBN 0-471-33061-2.
- ↑ Ziemer MA, Mason A, Carlson DM (September 1982). "Cell-free translations of proline-rich protein mRNAs". J. Biol. Chem. 257 (18): 11176–80. PMID 7107651. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=7107651.
- ↑ "OrthoMaM phylogenetic marker: TP53 coding sequence". http://www.orthomam.univ-montp2.fr/orthomam/data/cds/detailMarkers/ENSG00000141510_TP53.xml.
- ↑ Harms KL, Chen X (2005). "The C terminus of p53 family proteins is a cell fate determinant". Mol. Cell. Biol. 25 (5): 2014–30. doi:10.1128/MCB.25.5.2014-2030.2005. PMID 15713654.
- ↑ Piskacek S, Gregor M, Nemethova M, Grabner M, Kovarik P, Piskacek M (June 2007). "Nine-amino-acid transactivation domain: establishment and prediction utilities". Genomics 89 (6): 756–68. doi:10.1016/j.ygeno.2007.02.003. PMID 17467953.
- ↑ Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL (August 1997). "Induced alpha helix in the VP16 activation domain upon binding to a human TAF". Science (journal) 277 (5330): 1310–3. doi:10.1126/science.277.5330.1310. PMID 9271577.; Uesugi M, Verdine GL (December 1999). "The alpha-helical FXXPhiPhi motif in p53: TAF interaction and discrimination by MDM2". Proc. Natl. Acad. Sci. U.S.A. 96 (26): 14801–6. doi:10.1073/pnas.96.26.14801. PMID 10611293. PMC 24728. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=10611293.; Choi Y, Asada S, Uesugi M (May 2000). "Divergent hTAFII31-binding motifs hidden in activation domains". J. Biol. Chem. 275 (21): 15912–6. doi:10.1074/jbc.275.21.15912. PMID 10821850. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=10821850.; Venot C, Maratrat M, Sierra V, Conseiller E, Debussche L (April 1999). "Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains". Oncogene 18 (14): 2405–10. doi:10.1038/sj.onc.1202539. PMID 10327062.; Lin J, Chen J, Elenbaas B, Levine AJ (May 1994). "Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein". Genes Dev. 8 (10): 1235–46. doi:10.1101/gad.8.10.1235. PMID 7926727. http://www.genesdev.org/cgi/pmidlookup?view=long&pmid=7926727.
- ↑ Piskacek S, Gregor M, Nemethova M, Grabner M, Kovarik P, Piskacek M (June 2007). "Nine-amino-acid transactivation domain: establishment and prediction utilities". Genomics 89 (6): 756–68. doi:10.1016/j.ygeno.2007.02.003. PMID 17467953.; Piskacek M (2009-11-05). "9aaTAD is a common transactivation domain recruits multiple general coactivators TAF9, MED15, CBP/p300 and GCN5". Nature Precedings Pre-publication. doi:10.1038/npre.2009.3488.2.; Piskacek M (2009-11-05). "9aaTADs mimic DNA to interact with a pseudo-DNA Binding Domain KIX of Med15 (Molecular Chameleons)". Nature Precedings Pre-publication. doi:10.1038/npre.2009.3939.1.; Piskacek M; Piskacek, Martin (2009-11-20). "9aaTAD Prediction result (2006)". Nature Precedings Pre-publication. doi:10.1038/npre.2009.3984.1.
- ↑ The prediction for 9aaTADs (for both acidic and hydrophilic transactivation domains) is available online from ExPASy http://us.expasy.org/tools/ and EMBnet Spain http://www.es.embnet.org/Services/EMBnetAT/htdoc/9aatad/
- ↑ Bell S, Klein C, Müller L, Hansen S, Buchner J (2002). "p53 contains large unstructured regions in its native state". J. Mol. Biol. 322 (5): 917–27. doi:10.1016/S0022-2836(02)00848-3. PMID 12367518.
- ↑ National Center for Biotechnology Information. "The p53 tumor suppressor protein". Genes and Disease. United States National Institutes of Health. http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowSection&rid=gnd.section.107. Retrieved 2008-05-28.
- ↑ Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH (1998). "p14ARF links the tumour suppressors RB and p53". Nature 395 (6698): 124–5. doi:10.1038/25867. PMID 9744267.
- ↑ Wenwei Hu, Zhaohui Feng, Angelika K. Teresky1, Arnold J. Levine (November 29, 2007). "p53 regulates maternal reproduction through LIF". Nature 450, 721-724. http://www.nature.com/nature/journal/v450/n7170/abs/nature05993.html.
- ↑ "Genome's guardian gets a tan started". New Scientist. March 17, 2007. http://www.newscientist.com/channel/health/mg19325955.800-genomes-guardian-gets-a-tan-started.html. Retrieved 2007-03-29.
- ↑ Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D'Orazio J, Fung CY, Schanbacher CF, Granter SR, Fisher DE (2007). "Central role of p53 in the suntan response and pathologic hyperpigmentation". Cell 128 (5): 853–64. doi:10.1016/j.cell.2006.12.045. PMID 17350573.
- ↑ Han ES, Muller FL, Pérez VI, Qi W, Liang H, Xi L, Fu C, Doyle E, Hickey M, Cornell J, Epstein CJ, Roberts LJ, Van Remmen H, Richardson A (June 2008). "The in vivo gene expression signature of oxidative stress". Physiol. Genomics 34 (1): 112–26. doi:10.1152/physiolgenomics.00239.2007. PMID 18445702.
- ↑ Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober (March 2008). "Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice". Circ. Res. 102 (6): 703–10. doi:10.1161/CIRCRESAHA.107.164558. PMID 18239138.
- ↑ Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991). "p53 mutations in human cancers". Science 253 (5015): 49–53. doi:10.1126/science.1905840. PMID 1905840.
- ↑ Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee Park S, Thompson T, Karsenty G, Bradley A, Donehower LA (2002). "p53 mutant mice that display early ageing-associated phenotypes". Nature 415 (6867): 45–53. doi:10.1038/415045a. PMID 11780111.
- ↑ Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007). "Restoration of p53 function leads to tumour regression in vivo". Nature 445 (7128): 661–5. doi:10.1038/nature05541. PMID 17251932.
- ↑ Clemens A. Schmitt; Fridman, JS; Yang, M; Baranov, E; Hoffman, RM; Lowe, SW (April 2002). "Dissecting p53 tumor suppressor functions in vivo". Cancer Cell 1 (3): 289–298. doi:10.1016/S1535-6108(02)00047-8. PMID 12086865.
- ↑ Angeletti PC, Zhang L, Wood C (2008). "The viral etiology of AIDS-associated malignancies". Adv. Pharmacol. 56: 509–57. doi:10.1016/S1054-3589(07)56016-3. PMID 18086422.
- ↑ Chumakov P, Iotsova V, Georgiev G (1982). "Isolation of a plasmid clone containing the mRNA sequence for mouse nonviral T-antigen [Isolation of a plasmid clone containing the mRNA sequence for mouse nonviral T-antigen]". Doklady Akademii Nauk SSSR 267 (5): 1272–5. PMID 6295732.
- ↑ Oren M, Levine AJ (January 1983). "Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen". Proc. Natl. Acad. Sci. U.S.A. 80 (1): 56–9. doi:10.1073/pnas.80.1.56. PMID 6296874. PMC 393308. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=6296874.
- ↑ Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B (April 1989). "Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas". Science (journal) 244 (4901): 217–21. doi:10.1126/science.2649981. PMID 2649981.
- ↑ Maltzman W, Czyzyk L (1984). "UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells". Mol Cell Biol 4 (9): 1689–94. PMID 6092932.
- ↑ Kastan MB, Kuerbitz SJ (December 1993). "Control of G1 arrest after DNA damage". Environ. Health Perspect. (Environmental Health Perspectives, Vol. 101) 101 Suppl 5: 55–8. doi:10.2307/3431842. PMID 8013425. PMC 1519427. http://jstor.org/stable/3431842.
- ↑ Koshland DE (1993). "Molecule of the year". Science 262 (5142): 1953. doi:10.1126/science.8266084. PMID 8266084.
- ↑ Kondo, Seiji; Lu Ying, Debbas Michael, Lin Athena W, Sarosi Ildiko, Itie Annick, Wakeham Andrew, Tuan JoAnn, Saris Chris, Elliott Gary, Ma Weili, Benchimol Samuel, Lowe Scott W, Mak Tak Wah, Thukral Sushil K (Apr. 2003). "Characterization of cells and gene-targeted mice deficient for the p53-binding kinase homeodomain-interacting protein kinase 1 (HIPK1)". Proc. Natl. Acad. Sci. U.S.A. 100 (9): 5431–6. doi:10.1073/pnas.0530308100. PMID 12702766.
- ↑ Romanova, Larisa Y; Willers Henning, Blagosklonny Mikhail V, Powell Simon N (Dec. 2004). "The interaction of p53 with replication protein A mediates suppression of homologous recombination". Oncogene 23 (56): 9025–33. doi:10.1038/sj.onc.1207982. PMID 15489903.
- ↑ Riva, F; Zuco V, Vink A A, Supino R, Prosperi E (Dec. 2001). "UV-induced DNA incision and proliferating cell nuclear antigen recruitment to repair sites occur independently of p53-replication protein A interaction in p53 wild type and mutant ovarian carcinoma cells". Carcinogenesis 22 (12): 1971–8. doi:10.1093/carcin/22.12.1971. PMID 11751427.
- ↑ 34.0 34.1 Wang, X W; Yeh H, Schaeffer L, Roy R, Moncollin V, Egly J M, Wang Z, Freidberg E C, Evans M K, Taffe B G (Jun. 1995). "p53 modulation of TFIIH-associated nucleotide excision repair activity". Nat. Genet. 10 (2): 188–95. doi:10.1038/ng0695-188. PMID 7663514.
- ↑ Yu, A; Fan H Y, Liao D, Bailey A D, Weiner A M (May. 2000). "Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2, and 5S genes". Mol. Cell 5 (5): 801–10. doi:10.1016/S1097-2765(00)80320-2. PMID 10882116.
- ↑ Li, L; Liao J, Ruland J, Mak T W, Cohen S N (Feb. 2001). "A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control". Proc. Natl. Acad. Sci. U.S.A. 98 (4): 1619–24. doi:10.1073/pnas.98.4.1619. PMID 11172000.
- ↑ Cuddihy, A R; Wong A H, Tam N W, Li S, Koromilas A E (Apr. 1999). "The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro". Oncogene 18 (17): 2690–702. doi:10.1038/sj.onc.1202620. PMID 10348343.
- ↑ Abedini, Mohammad R; Muller Emilie J, Brun Jan, Bergeron Richard, Gray Douglas A, Tsang Benjamin K (Jun. 2008). "Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells". Cancer Res. 68 (12): 4511–7. doi:10.1158/0008-5472.CAN-08-0673. PMID 18559494.
- ↑ 39.0 39.1 Ito, Akihiro; Kawaguchi Yoshiharu, Lai Chun-Hsiang, Kovacs Jeffrey J, Higashimoto Yuichiro, Appella Ettore, Yao Tso-Pang (Nov. 2002). "MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation". EMBO J. 21 (22): 6236–45. doi:10.1093/emboj/cdf616. PMID 12426395.
- ↑ 40.0 40.1 Livengood, Jill A; Scoggin Kirsten E S, Van Orden Karen, McBryant Steven J, Edayathumangalam Rajeswari S, Laybourn Paul J, Nyborg Jennifer K (Mar. 2002). "p53 Transcriptional activity is mediated through the SRC1-interacting domain of CBP/p300". J. Biol. Chem. 277 (11): 9054–61. doi:10.1074/jbc.M108870200. PMID 11782467.
- ↑ 41.0 41.1 Giebler, H A; Lemasson I, Nyborg J K (Jul. 2000). "p53 recruitment of CREB binding protein mediated through phosphorylated CREB: a novel pathway of tumor suppressor regulation". Mol. Cell. Biol. 20 (13): 4849–58. doi:10.1128/MCB.20.13.4849-4858.2000. PMID 10848610.
- ↑ Hu, M C; Qiu W R, Wang Y P (Nov. 1997). "JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases". Oncogene 15 (19): 2277–87. doi:10.1038/sj.onc.1201401. PMID 9393873.
- ↑ Lin, Yenshou; Khokhlatchev Andrei, Figeys Daniel, Avruch Joseph (Dec. 2002). "Death-associated protein 4 binds MST1 and augments MST1-induced apoptosis". J. Biol. Chem. 277 (50): 47991–8001. doi:10.1074/jbc.M202630200. PMID 12384512.
- ↑ Fusaro, Gina; Dasgupta Piyali, Rastogi Shipra, Joshi Bharat, Chellappan Srikumar (Nov. 2003). "Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling". J. Biol. Chem. 278 (48): 47853–61. doi:10.1074/jbc.M305171200. PMID 14500729.
- ↑ 45.0 45.1 45.2 Zhang, Yanping; Wolf Gabrielle White, Bhat Krishna, Jin Aiwen, Allio Theresa, Burkhart William A, Xiong Yue (Dec. 2003). "Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway". Mol. Cell. Biol. 23 (23): 8902–12. doi:10.1128/MCB.23.23.8902-8912.2003. PMID 14612427.
- ↑ Kang, Jian; Ferguson David, Song Hoseok, Bassing Craig, Eckersdorff Mark, Alt Frederick W, Xu Yang (Jan. 2005). "Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression". Mol. Cell. Biol. 25 (2): 661–70. doi:10.1128/MCB.25.2.661-670.2005. PMID 15632067.
- ↑ 47.0 47.1 Fabbro, Megan; Savage Kienan, Hobson Karen, Deans Andrew J, Powell Simon N, McArthur Grant A, Khanna Kum Kum (Jul. 2004). "BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage". J. Biol. Chem. 279 (30): 31251–8. doi:10.1074/jbc.M405372200. PMID 15159397.
- ↑ Khanna, K K; Keating K E, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller S P, Lavin M F (Dec. 1998). "ATM associates with and phosphorylates p53: mapping the region of interaction". Nat. Genet. 20 (4): 398–400. doi:10.1038/3882. PMID 9843217.
- ↑ Westphal, C H; Schmaltz C, Rowan S, Elson A, Fisher D E, Leder P (May. 1997). "Genetic interactions between atm and p53 influence cellular proliferation and irradiation-induced cell cycle checkpoints". Cancer Res. 57 (9): 1664–7. PMID 9135004.
- ↑ 50.0 50.1 50.2 Kim, S T; Lim D S, Canman C E, Kastan M B (Dec. 1999). "Substrate specificities and identification of putative substrates of ATM kinase family members". J. Biol. Chem. 274 (53): 37538–43. doi:10.1074/jbc.274.53.37538. PMID 10608806.
- ↑ 51.0 51.1 Goudelock, Dawn Marie; Jiang Kecheng, Pereira Elizabeth, Russell Beatriz, Sanchez Yolanda (Aug. 2003). "Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex". J. Biol. Chem. 278 (32): 29940–7. doi:10.1074/jbc.M301765200. PMID 12756247.
- ↑ Yavuzer, U; Smith G C, Bliss T, Werner D, Jackson S P (Jul. 1998). "DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D". Genes Dev. 12 (14): 2188–99. doi:10.1101/gad.12.14.2188. PMID 9679063.
- ↑ Stelzl, Ulrich; Worm Uwe, Lalowski Maciej, Haenig Christian, Brembeck Felix H, Goehler Heike, Stroedicke Martin, Zenkner Martina, Schoenherr Anke, Koeppen Susanne, Timm Jan, Mintzlaff Sascha, Abraham Claudia, Bock Nicole, Kietzmann Silvia, Goedde Astrid, Toksöz Engin, Droege Anja, Krobitsch Sylvia, Korn Bernhard, Birchmeier Walter, Lehrach Hans, Wanker Erich E (Sep. 2005). "A human protein-protein interaction network: a resource for annotating the proteome". Cell 122 (6): 957–68. doi:10.1016/j.cell.2005.08.029. PMID 16169070.
- ↑ Yan, Chunhong; Wang Heng, Boyd Douglas D (Mar. 2002). "ATF3 represses 72-kDa type IV collagenase (MMP-2) expression by antagonizing p53-dependent trans-activation of the collagenase promoter". J. Biol. Chem. 277 (13): 10804–12. doi:10.1074/jbc.M112069200. PMID 11792711.
- ↑ Hofmann, Thomas G; Möller Andreas, Sirma Hüaeyin, Zentgraf Hanswalter, Taya Yoichi, Dröge Wulf, Will Hans, Schmitz M Lienhard (Jan. 2002). "Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2". Nat. Cell Biol. 4 (1): 1–10. doi:10.1038/ncb715. PMID 11740489.
- ↑ Kim, Eun-Joo; Park Jong-Sup, Um Soo-Jong (Aug. 2002). "Identification and characterization of HIPK2 interacting with p73 and modulating functions of the p53 family in vivo". J. Biol. Chem. 277 (35): 32020–8. doi:10.1074/jbc.M200153200. PMID 11925430.
- ↑ Chen, Delin; Li Muyang, Luo Jianyuan, Gu Wei (Apr. 2003). "Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function". J. Biol. Chem. 278 (16): 13595–8. doi:10.1074/jbc.C200694200. PMID 12606552.
- ↑ Ravi, R; Mookerjee B, Bhujwalla Z M, Sutter C H, Artemov D, Zeng Q, Dillehay L E, Madan A, Semenza G L, Bedi A (Jan. 2000). "Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha". Genes Dev. 14 (1): 34–44. PMID 10640274.
- ↑ Hansson, Lars O; Friedler Assaf, Freund Stefan, Rudiger Stefan, Fersht Alan R (Aug. 2002). "Two sequence motifs from HIF-1alpha bind to the DNA-binding site of p53". Proc. Natl. Acad. Sci. U.S.A. 99 (16): 10305–9. doi:10.1073/pnas.122347199. PMID 12124396.
- ↑ An, W G; Kanekal M, Simon M C, Maltepe E, Blagosklonny M V, Neckers L M (Mar. 1998). "Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha". Nature 392 (6674): 405–8. doi:10.1038/32925. PMID 9537326.
- ↑ Frade, R; Balbo M, Barel M (Dec. 2000). "RB18A, whose gene is localized on chromosome 17q12-q21.1, regulates in vivo p53 transactivating activity". Cancer Res. 60 (23): 6585–9. PMID 11118038.
- ↑ Drané, P; Barel M, Balbo M, Frade R (Dec. 1997). "Identification of RB18A, a 205 kDa new p53 regulatory protein which shares antigenic and functional properties with p53". Oncogene 15 (25): 3013–24. doi:10.1038/sj.onc.1201492. PMID 9444950.
- ↑ Liu, J; Grogan L, Nau M M, Allegra C J, Chu E, Wright J J (Apr. 2001). "Physical interaction between p53 and primary response gene Egr-1". Int. J. Oncol. 18 (4): 863–70. PMID 11251186.
- ↑ Peng, Y C; Kuo F, Breiding D E, Wang Y F, Mansur C P, Androphy E J (Sep. 2001). "AMF1 (GPS2) modulates p53 transactivation". Mol. Cell. Biol. 21 (17): 5913–24. doi:10.1128/MCB.21.17.5913-5924.2001. PMID 11486030.
- ↑ 65.0 65.1 65.2 Gallagher, W M; Argentini M, Sierra V, Bracco L, Debussche L, Conseiller E (Jun. 1999). "MBP1: a novel mutant p53-specific protein partner with oncogenic properties". Oncogene 18 (24): 3608–16. doi:10.1038/sj.onc.1202937. PMID 10380882.
- ↑ 66.0 66.1 Kurki, Sari; Latonen Leena, Laiho Marikki (Oct. 2003). "Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization". J. Cell. Sci. 116 (Pt 19): 3917–25. doi:10.1242/jcs.00714. PMID 12915590.
- ↑ Fogal, V; Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi P P, Will H, Schneider C, Del Sal G (Nov. 2000). "Regulation of p53 activity in nuclear bodies by a specific PML isoform". EMBO J. 19 (22): 6185–95. doi:10.1093/emboj/19.22.6185. PMID 11080164.
- ↑ Guo, A; Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi P P (Oct. 2000). "The function of PML in p53-dependent apoptosis". Nat. Cell Biol. 2 (10): 730–6. doi:10.1038/35036365. PMID 11025664.
- ↑ Xie, S; Wu H, Wang Q, Cogswell J P, Husain I, Conn C, Stambrook P, Jhanwar-Uniyal M, Dai W (Nov. 2001). "Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway". J. Biol. Chem. 276 (46): 43305–12. doi:10.1074/jbc.M106050200. PMID 11551930.
- ↑ Bahassi, El Mustapha; Conn Christopher W, Myer David L, Hennigan Robert F, McGowan Clare H, Sanchez Yolanda, Stambrook Peter J (Sep. 2002). "Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways". Oncogene 21 (43): 6633–40. doi:10.1038/sj.onc.1205850. PMID 12242661.
- ↑ Tomasini, Richard; Samir Amina Azizi, Carrier Alice, Isnardon Daniel, Cecchinelli Barbara, Soddu Silvia, Malissen Bernard, Dagorn Jean-Charles, Iovanna Juan L, Dusetti Nelson J (Sep. 2003). "TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity". J. Biol. Chem. 278 (39): 37722–9. doi:10.1074/jbc.M301979200. PMID 12851404.
- ↑ Okamura, S; Arakawa H, Tanaka T, Nakanishi H, Ng C C, Taya Y, Monden M, Nakamura Y (Jul. 2001). "p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis". Mol. Cell 8 (1): 85–94. doi:10.1016/S1097-2765(01)00284-2. PMID 11511362.
- ↑ Chen, Shih-Shun; Chang Pi-Chu, Cheng Yu-Wen, Tang Fen-Mei, Lin Young-Sun (Sep. 2002). "Suppression of the STK15 oncogenic activity requires a transactivation-independent p53 function". EMBO J. 21 (17): 4491–9. doi:10.1093/emboj/cdf409. PMID 12198151.
- ↑ 74.0 74.1 74.2 74.3 74.4 74.5 Dong, Yuanshu; Hakimi Mohamed-Ali, Chen Xiaowei, Kumaraswamy Easwari, Cooch Neil S, Godwin Andrew K, Shiekhattar Ramin (Nov. 2003). "Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair". Mol. Cell 12 (5): 1087–99. doi:10.1016/S1097-2765(03)00424-6. PMID 14636569.
- ↑ Bernal, Juan A; Luna Rosa, Espina Agueda, Lázaro Icíar, Ramos-Morales Francisco, Romero Francisco, Arias Carmen, Silva Augusto, Tortolero María, Pintor-Toro José A (Oct. 2002). "Human securin interacts with p53 and modulates p53-mediated transcriptional activity and apoptosis". Nat. Genet. 32 (2): 306–11. doi:10.1038/ng997. PMID 12355087.
- ↑ Tian, Hui; Faje Alexander T, Lee Siu Lan, Jorgensen Timothy J (2002). "Radiation-induced phosphorylation of Chk1 at S345 is associated with p53-dependent cell cycle arrest pathways". Neoplasia 4 (2): 171–80. doi:10.1038/sj/neo/7900219. PMID 11896572.
- ↑ 77.0 77.1 77.2 Sengupta, Sagar; Robles Ana I, Linke Steven P, Sinogeeva Natasha I, Zhang Ran, Pedeux Remy, Ward Irene M, Celeste Arkady, Nussenzweig André, Chen Junjie, Halazonetis Thanos D, Harris Curtis C (Sep. 2004). "Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest". J. Cell Biol. 166 (6): 801–13. doi:10.1083/jcb.200405128. PMID 15364958.
- ↑ Zhao, Lili; Samuels Tina, Winckler Sarah, Korgaonkar Chandrashekhar, Tompkins Van, Horne Mary C, Quelle Dawn E (Jan. 2003). "Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways". Mol. Cancer Res. 1 (3): 195–206. PMID 12556559.
- ↑ 79.0 79.1 Schneider, E; Montenarh M, Wagner P (Nov. 1998). "Regulation of CAK kinase activity by p53". Oncogene 17 (21): 2733–41. doi:10.1038/sj.onc.1202504. PMID 9840937.
- ↑ Gobert, C; Skladanowski A, Larsen A K (Aug. 1999). "The interaction between p53 and DNA topoisomerase I is regulated differently in cells with wild-type and mutant p53". Proc. Natl. Acad. Sci. U.S.A. 96 (18): 10355–60. doi:10.1073/pnas.96.18.10355. PMID 10468612.
- ↑ Mao, Yinghui; Mehl Issac R, Muller Mark T (Feb. 2002). "Subnuclear distribution of topoisomerase I is linked to ongoing transcription and p53 status". Proc. Natl. Acad. Sci. U.S.A. 99 (3): 1235–40. doi:10.1073/pnas.022631899. PMID 11805286.
- ↑ Kahyo, T; Nishida T, Yasuda H (Sep. 2001). "Involvement of PIAS1 in the sumoylation of tumor suppressor p53". Mol. Cell 8 (3): 713–8. doi:10.1016/S1097-2765(01)00349-5. PMID 11583632.
- ↑ 83.0 83.1 Li, L; Ljungman M, Dixon J E (Jan. 2000). "The human Cdc14 phosphatases interact with and dephosphorylate the tumor suppressor protein p53". J. Biol. Chem. 275 (4): 2410–4. doi:10.1074/jbc.275.4.2410. PMID 10644693.
- ↑ Marmorstein, L Y; Ouchi T, Aaronson S A (Nov. 1998). "The BRCA2 gene product functionally interacts with p53 and RAD51". Proc. Natl. Acad. Sci. U.S.A. 95 (23): 13869–74. doi:10.1073/pnas.95.23.13869. PMID 9811893.
- ↑ Abramovitch, S; Werner H (2003). "Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene". Horm. Metab. Res. 35 (11-12): 758–62. doi:10.1055/s-2004-814154. PMID 14710355.
- ↑ Ouchi, T; Monteiro A N, August A, Aaronson S A, Hanafusa H (Mar. 1998). "BRCA1 regulates p53-dependent gene expression". Proc. Natl. Acad. Sci. U.S.A. 95 (5): 2302–6. doi:10.1073/pnas.95.5.2302. PMID 9482880.
- ↑ Chai, Y L; Cui J, Shao N, Shyam E, Reddy P, Rao V N (Jan. 1999). "The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter". Oncogene 18 (1): 263–8. doi:10.1038/sj.onc.1202323. PMID 9926942.
- ↑ Zhang, H; Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, Weber B L, El-Deiry W S (Apr. 1998). "BRCA1 physically associates with p53 and stimulates its transcriptional activity". Oncogene 16 (13): 1713–21. doi:10.1038/sj.onc.1201932. PMID 9582019.
- ↑ Leng, Roger P; Lin Yunping, Ma Weili, Wu Hong, Lemmers Benedicte, Chung Stephen, Parant John M, Lozano Guillermina, Hakem Razqallah, Benchimol Samuel (Mar. 2003). "Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation". Cell 112 (6): 779–91. doi:10.1016/S0092-8674(03)00193-4. PMID 12654245.
- ↑ Sheng, Yi; Laister Rob C, Lemak Alexander, Wu Bin, Tai Elizabeth, Duan Shili, Lukin Jonathan, Sunnerhagen Maria, Srisailam Sampath, Karra Murthy, Benchimol Sam, Arrowsmith Cheryl H (Dec. 2008). "Molecular basis of Pirh2-mediated p53 ubiquitylation". Nat. Struct. Mol. Biol. 15 (12): 1334–42. doi:10.1038/nsmb.1521. PMID 19043414.
- ↑ 91.0 91.1 Rizos, Helen; Diefenbach Eve, Badhwar Prerna, Woodruff Sarah, Becker Therese M, Rooney Robert J, Kefford Richard F (Feb. 2003). "Association of p14ARF with the p120E4F transcriptional repressor enhances cell cycle inhibition". J. Biol. Chem. 278 (7): 4981–9. doi:10.1074/jbc.M210978200. PMID 12446718.
- ↑ Sandy, P; Gostissa M, Fogal V, Cecco L D, Szalay K, Rooney R J, Schneider C, Del Sal G (Jan. 2000). "p53 is involved in the p120E4F-mediated growth arrest". Oncogene 19 (2): 188–99. doi:10.1038/sj.onc.1203250. PMID 10644996.
- ↑ 93.0 93.1 Gueven, Nuri; Becherel Olivier J, Kijas Amanda W, Chen Philip, Howe Orla, Rudolph Jeanette H, Gatti Richard, Date Hidetoshi, Onodera Osamu, Taucher-Scholz Gisela, Lavin Martin F (May. 2004). "Aprataxin, a novel protein that protects against genotoxic stress". Hum. Mol. Genet. 13 (10): 1081–93. doi:10.1093/hmg/ddh122. PMID 15044383.
- ↑ Malanga, M; Pleschke J M, Kleczkowska H E, Althaus F R (May. 1998). "Poly(ADP-ribose) binds to specific domains of p53 and alters its DNA binding functions". J. Biol. Chem. 273 (19): 11839–43. doi:10.1074/jbc.273.19.11839. PMID 9565608.
- ↑ Bai, L; Merchant J L (Jul. 2001). "ZBP-89 promotes growth arrest through stabilization of p53". Mol. Cell. Biol. 21 (14): 4670–83. doi:10.1128/MCB.21.14.4670-4683.2001. PMID 11416144.
- ↑ Imamura, T; Izumi H, Nagatani G, Ise T, Nomoto M, Iwamoto Y, Kohno K (Mar. 2001). "Interaction with p53 enhances binding of cisplatin-modified DNA by high mobility group 1 protein". J. Biol. Chem. 276 (10): 7534–40. doi:10.1074/jbc.M008143200. PMID 11106654.
- ↑ Dintilhac, Agnès; Bernués Jordi (Mar. 2002). "HMGB1 interacts with many apparently unrelated proteins by recognizing short amino acid sequences". J. Biol. Chem. 277 (9): 7021–8. doi:10.1074/jbc.M108417200. PMID 11748221.
- ↑ 98.0 98.1 Han, Jung Min; Park Bum-Joon, Park Sang Gyu, Oh Young Sun, Choi So Jung, Lee Sang Won, Hwang Soon-Kyung, Chang Seung-Hee, Cho Myung-Haing, Kim Sunghoon (Aug. 2008). "AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stresses via p53". Proc. Natl. Acad. Sci. U.S.A. 105 (32): 11206–11. doi:10.1073/pnas.0800297105. PMID 18695251.
- ↑ Simons, A; Melamed-Bessudo C, Wolkowicz R, Sperling J, Sperling R, Eisenbach L, Rotter V (Jan. 1997). "PACT: cloning and characterization of a cellular p53 binding protein that interacts with Rb". Oncogene 14 (2): 145–55. doi:10.1038/sj.onc.1200825. PMID 9010216.
- ↑ Derbyshire, Dean J; Basu Balaku P, Serpell Louise C, Joo Woo S, Date Takayasu, Iwabuchi Kuniyoshi, Doherty Aidan J (Jul. 2002). "Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor". EMBO J. 21 (14): 3863–72. doi:10.1093/emboj/cdf383. PMID 12110597.
- ↑ Ekblad, Caroline M S; Friedler Assaf, Veprintsev Dmitry, Weinberg Richard L, Itzhaki Laura S (Mar. 2004). "Comparison of BRCT domains of BRCA1 and 53BP1: a biophysical analysis". Protein Sci. 13 (3): 617–25. doi:10.1110/ps.03461404. PMID 14978302.
- ↑ Lo, Kevin W-H; Kan Ho-Man, Chan Ling-Nga, Xu Wei-Guang, Wang Ke-Peng, Wu Zhenguo, Sheng Morgan, Zhang Mingjie (Mar. 2005). "The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation". J. Biol. Chem. 280 (9): 8172–9. doi:10.1074/jbc.M411408200. PMID 15611139.
- ↑ Joo, Woo S; Jeffrey Philip D, Cantor Sharon B, Finnin Michael S, Livingston David M, Pavletich Nikola P (Mar. 2002). "Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure". Genes Dev. 16 (5): 583–93. doi:10.1101/gad.959202. PMID 11877378.
- ↑ Derbyshire, Dean J; Basu Balaku P, Date Takayasu, Iwabuchi Kuniyoshi, Doherty Aidan J (Oct. 2002). "Purification, crystallization and preliminary X-ray analysis of the BRCT domains of human 53BP1 bound to the p53 tumour suppressor". Acta Crystallogr. D Biol. Crystallogr. 58 (Pt 10 Pt 2): 1826–9. doi:10.1107/S0907444902010910. PMID 12351827.
- ↑ 105.0 105.1 Iwabuchi, K; Bartel P L, Li B, Marraccino R, Fields S (Jun. 1994). "Two cellular proteins that bind to wild-type but not mutant p53". Proc. Natl. Acad. Sci. U.S.A. 91 (13): 6098–102. doi:10.1073/pnas.91.13.6098. PMID 8016121.
- ↑ Luciani, M G; Hutchins J R, Zheleva D, Hupp T R (Jul. 2000). "The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A". J. Mol. Biol. 300 (3): 503–18. doi:10.1006/jmbi.2000.3830. PMID 10884347.
- ↑ Ababneh, M; Götz C, Montenarh M (May. 2001). "Downregulation of the cdc2/cyclin B protein kinase activity by binding of p53 to p34(cdc2)". Biochem. Biophys. Res. Commun. 283 (2): 507–12. doi:10.1006/bbrc.2001.4792. ISSN 0006-291X. PMID 11327730.
- ↑ Naumovski, L; Cleary M L (Jul. 1996). "The p53-binding protein 53BP2 also interacts with Bc12 and impedes cell cycle progression at G2/M". Mol. Cell. Biol. 16 (7): 3884–92. PMID 8668206.
- ↑ 109.0 109.1 Cowell, I G; Okorokov A L, Cutts S A, Padget K, Bell M, Milner J, Austin C A (Feb. 2000). "Human topoisomerase IIalpha and IIbeta interact with the C-terminal region of p53". Exp. Cell Res. 255 (1): 86–94. doi:10.1006/excr.1999.4772. PMID 10666337.
- ↑ Wang, X W; Tseng A, Ellis N A, Spillare E A, Linke S P, Robles A I, Seker H, Yang Q, Hu P, Beresten S, Bemmels N A, Garfield S, Harris C C (Aug. 2001). "Functional interaction of p53 and BLM DNA helicase in apoptosis". J. Biol. Chem. 276 (35): 32948–55. doi:10.1074/jbc.M103298200. PMID 11399766.
- ↑ Garkavtsev, I V; Kley N, Grigorian I A, Gudkov A V (Dec. 2001). "The Bloom syndrome protein interacts and cooperates with p53 in regulation of transcription and cell growth control". Oncogene 20 (57): 8276–80. doi:10.1038/sj.onc.1205120. PMID 11781842.
- ↑ 112.0 112.1 Yang, Qin; Zhang Ran, Wang Xin Wei, Spillare Elisa A, Linke Steven P, Subramanian Deepa, Griffith Jack D, Li Ji Liang, Hickson Ian D, Shen Jiang Cheng, Loeb Lawrence A, Mazur Sharlyn J, Appella Ettore, Brosh Robert M, Karmakar Parimal, Bohr Vilhelm A, Harris Curtis C (Aug. 2002). "The processing of Holliday junctions by BLM and WRN helicases is regulated by p53". J. Biol. Chem. 277 (35): 31980–7. doi:10.1074/jbc.M204111200. PMID 12080066.
- ↑ Leu, J I-Ju; Dumont Patrick, Hafey Michael, Murphy Maureen E, George Donna L (May. 2004). "Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex". Nat. Cell Biol. 6 (5): 443–50. doi:10.1038/ncb1123. PMID 15077116.
- ↑ Nikolaev, Anatoly Y; Li Muyang, Puskas Norbert, Qin Jun, Gu Wei (Jan. 2003). "Parc: a cytoplasmic anchor for p53". Cell 112 (1): 29–40. doi:10.1016/S0092-8674(02)01255-2. PMID 12526791.
- ↑ Stürzbecher, H W; Donzelmann B, Henning W, Knippschild U, Buchhop S (Apr. 1996). "p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction". EMBO J. 15 (8): 1992–2002. PMID 8617246.
- ↑ Buchhop, S; Gibson M K, Wang X W, Wagner P, Stürzbecher H W, Harris C C (Oct. 1997). "Interaction of p53 with the human Rad51 protein". Nucleic Acids Res. 25 (19): 3868–74. doi:10.1093/nar/25.19.3868. PMID 9380510.
- ↑ 117.0 117.1 117.2 Zhang, Zhuo; Zhang Ruiwen (Mar. 2008). "Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation". EMBO J. 27 (6): 852–64. doi:10.1038/emboj.2008.25. PMID 18309296.
- ↑ Lin, Jing; Yang Qingyuan, Yan Zhe, Markowitz Joseph, Wilder Paul T, Carrier France, Weber David J (Aug. 2004). "Inhibiting S100B restores p53 levels in primary malignant melanoma cancer cells". J. Biol. Chem. 279 (32): 34071–7. doi:10.1074/jbc.M405419200. PMID 15178678.
- ↑ 119.0 119.1 Minty, A; Dumont X, Kaghad M, Caput D (Nov. 2000). "Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif". J. Biol. Chem. 275 (46): 36316–23. doi:10.1074/jbc.M004293200. PMID 10961991.
- ↑ Shen, Z; Pardington-Purtymun P E, Comeaux J C, Moyzis R K, Chen D J (Oct. 1996). "Associations of UBE2I with RAD52, UBL1, p53, and RAD51 proteins in a yeast two-hybrid system". Genomics 37 (2): 183–6. doi:10.1006/geno.1996.0540. PMID 8921390.
- ↑ Bernier-Villamor, Victor; Sampson Deborah A, Matunis Michael J, Lima Christopher D (Feb. 2002). "Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1". Cell 108 (3): 345–56. doi:10.1016/S0092-8674(02)00630-X. PMID 11853669.
- ↑ Wulf, Gerburg M; Liou Yih-Cherng, Ryo Akihide, Lee Sam W, Lu Kun Ping (Dec. 2002). "Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage". J. Biol. Chem. 277 (50): 47976–9. doi:10.1074/jbc.C200538200. PMID 12388558.
- ↑ Zacchi, Paola; Gostissa Monica, Uchida Takafumi, Salvagno Clio, Avolio Fabio, Volinia Stefano, Ronai Ze'ev, Blandino Giovanni, Schneider Claudio, Del Sal Giannino (Oct. 2002). "The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults". Nature 419 (6909): 853–7. doi:10.1038/nature01120. PMID 12397362.
- ↑ 124.0 124.1 Ivanchuk, Stacey M; Mondal Soma, Rutka James T (Jun. 2008). "p14ARF interacts with DAXX: effects on HDM2 and p53". Cell Cycle 7 (12): 1836–50. PMID 18583933.
- ↑ Steffan, J S; Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu Y Z, Gohler H, Wanker E E, Bates G P, Housman D E, Thompson L M (Jun. 2000). "The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription". Proc. Natl. Acad. Sci. U.S.A. 97 (12): 6763–8. doi:10.1073/pnas.100110097. PMID 10823891.
- ↑ 126.0 126.1 Freeman, Daniel J; Li Andrew G, Wei Gang, Li Heng-Hong, Kertesz Nathalie, Lesche Ralf, Whale Andrew D, Martinez-Diaz Hilda, Rozengurt Nora, Cardiff Robert D, Liu Xuan, Wu Hong (Feb. 2003). "PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms". Cancer Cell 3 (2): 117–30. doi:10.1016/S1535-6108(03)00021-7. PMID 12620407.
- ↑ 127.0 127.1 Akakura, S; Yoshida M, Yoneda Y, Horinouchi S (May. 2001). "A role for Hsc70 in regulating nucleocytoplasmic transport of a temperature-sensitive p53 (p53Val-135)". J. Biol. Chem. 276 (18): 14649–57. doi:10.1074/jbc.M100200200. PMID 11297531.
- ↑ 128.0 128.1 Ko, L J; Shieh S Y, Chen X, Jayaraman L, Tamai K, Taya Y, Prives C, Pan Z Q (Dec. 1997). "p53 is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner". Mol. Cell. Biol. 17 (12): 7220–9. PMID 9372954.
- ↑ 129.0 129.1 Shiseki, Masayuki; Nagashima Makoto, Pedeux Remy M, Kitahama-Shiseki Mariko, Miura Koh, Okamura Shu, Onogi Hitoshi, Higashimoto Yuichiro, Appella Ettore, Yokota Jun, Harris Curtis C (May. 2003). "p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity". Cancer Res. 63 (10): 2373–8. PMID 12750254.
- ↑ Shinobu, N; Maeda T, Aso T, Ito T, Kondo T, Koike K, Hatakeyama M (Jun. 1999). "Physical interaction and functional antagonism between the RNA polymerase II elongation factor ELL and p53". J. Biol. Chem. 274 (24): 17003–10. doi:10.1074/jbc.274.24.17003. PMID 10358050.
- ↑ Lim, Ssang-Taek; Chen Xiao Lei, Lim Yangmi, Hanson Dan A, Vo Thanh-Trang, Howerton Kyle, Larocque Nicholas, Fisher Susan J, Schlaepfer David D, Ilic Dusko (Jan. 2008). "Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation". Mol. Cell 29 (1): 9–22. doi:10.1016/j.molcel.2007.11.031. PMID 18206965.
- ↑ 132.0 132.1 Colaluca, Ivan N; Tosoni Daniela, Nuciforo Paolo, Senic-Matuglia Francesca, Galimberti Viviana, Viale Giuseppe, Pece Salvatore, Di Fiore Pier Paolo (Jan. 2008). "NUMB controls p53 tumour suppressor activity". Nature 451 (7174): 76–80. doi:10.1038/nature06412. PMID 18172499.
- ↑ 133.0 133.1 Leung, Ka Man; Po Lai See, Tsang Fan Cheung, Siu Wai Yi, Lau Anita, Ho Horace T B, Poon Randy Y C (Sep. 2002). "The candidate tumor suppressor ING1b can stabilize p53 by disrupting the regulation of p53 by MDM2". Cancer Res. 62 (17): 4890–3. PMID 12208736.
- ↑ Garkavtsev, I; Grigorian I A, Ossovskaya V S, Chernov M V, Chumakov P M, Gudkov A V (Jan. 1998). "The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control". Nature 391 (6664): 295–8. doi:10.1038/34675. PMID 9440695.
- ↑ 135.0 135.1 Zhang, Y; Xiong Y, Yarbrough W G (Mar. 1998). "ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways". Cell 92 (6): 725–34. doi:10.1016/S0092-8674(00)81401-4. PMID 9529249.
- ↑ Li, Muyang; Chen Delin, Shiloh Ariel, Luo Jianyuan, Nikolaev Anatoly Y, Qin Jun, Gu Wei (Apr. 2002). "Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization". Nature 416 (6881): 648–53. doi:10.1038/nature737. PMID 11923872.
- ↑ Tsai, Kuo-Wang; Tseng Hsiao-Chun, Lin Wen-Chang (Oct. 2008). "Two wobble-splicing events affect ING4 protein subnuclear localization and degradation". Exp. Cell Res. 314 (17): 3130–41. doi:10.1016/j.yexcr.2008.08.002. PMID 18775696.
- ↑ Waterman, M J; Stavridi E S, Waterman J L, Halazonetis T D (Jun. 1998). "ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins". Nat. Genet. 19 (2): 175–8. doi:10.1038/542. PMID 9620776.
- ↑ Lyakhovich, Alex; Shekhar Malathy P V (Apr. 2003). "Supramolecular complex formation between Rad6 and proteins of the p53 pathway during DNA damage-induced response". Mol. Cell. Biol. 23 (7): 2463–75. doi:10.1128/MCB.23.7.2463-2475.2003. PMID 12640129.
- ↑ Sehat, Bita; Andersson Sandra, Girnita Leonard, Larsson Olle (Jul. 2008). "Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis". Cancer Res. 68 (14): 5669–77. doi:10.1158/0008-5472.CAN-07-6364. PMID 18632619.
- ↑ Song, Min Sup; Song Su Jung, Kim So Yeon, Oh Hyun Jung, Lim Dae-Sik (Jul. 2008). "The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex". EMBO J. 27 (13): 1863–74. doi:10.1038/emboj.2008.115. PMID 18566590.
- ↑ Yang, Wensheng; Dicker David T, Chen Jiandong, El-Deiry Wafik S (Mar. 2008). "CARPs enhance p53 turnover by degrading 14-3-3sigma and stabilizing MDM2". Cell Cycle 7 (5): 670–82. PMID 18382127.
- ↑ Abe, Yoshinori; Oda-Sato Eri, Tobiume Kei, Kawauchi Keiko, Taya Yoichi, Okamoto Koji, Oren Moshe, Tanaka Nobuyuki (Mar. 2008). "Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2". Proc. Natl. Acad. Sci. U.S.A. 105 (12): 4838–43. doi:10.1073/pnas.0712216105. PMID 18359851.
- ↑ Dohmesen, Christoph; Koeppel Max, Dobbelstein Matthias (Jan. 2008). "Specific inhibition of Mdm2-mediated neddylation by Tip60". Cell Cycle 7 (2): 222–31. PMID 18264029.
- ↑ Brosh, R M; Karmakar P, Sommers J A, Yang Q, Wang X W, Spillare E A, Harris C C, Bohr V A (Sep. 2001). "p53 Modulates the exonuclease activity of Werner syndrome protein". J. Biol. Chem. 276 (37): 35093–102. doi:10.1074/jbc.M103332200. PMID 11427532.
- ↑ Tsai, Robert Y L; McKay Ronald D G (Dec. 2002). "A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells". Genes Dev. 16 (23): 2991–3003. doi:10.1101/gad.55671. PMID 12464630.
- ↑ Uramoto, Hidetaka; Izumi Hiroto, Nagatani Gunji, Ohmori Haruki, Nagasue Naofumi, Ise Tomoko, Yoshida Takeshi, Yasumoto Kosei, Kohno Kimitoshi (Apr. 2003). "Physical interaction of tumour suppressor p53/p73 with CCAAT-binding transcription factor 2 (CTF2) and differential regulation of human high-mobility group 1 (HMG1) gene expression". Biochem. J. 371 (Pt 2): 301–10. doi:10.1042/BJ20021646. PMID 12534345.
- ↑ Chang, N S; Pratt N, Heath J, Schultz L, Sleve D, Carey G B, Zevotek N (Feb. 2001). "Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity". J. Biol. Chem. 276 (5): 3361–70. doi:10.1074/jbc.M007140200. PMID 11058590.
- ↑ 149.0 149.1 Kojic, Snezana; Medeot Elisa, Guccione Ernesto, Krmac Helena, Zara Ivano, Martinelli Valentina, Valle Giorgio, Faulkner Georgine (May. 2004). "The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle". J. Mol. Biol. 339 (2): 313–25. doi:10.1016/j.jmb.2004.03.071. PMID 15136035.
- ↑ Okamoto, T; Izumi H, Imamura T, Takano H, Ise T, Uchiumi T, Kuwano M, Kohno K (Dec. 2000). "Direct interaction of p53 with the Y-box binding protein, YB-1: a mechanism for regulation of human gene expression". Oncogene 19 (54): 6194–202. doi:10.1038/sj.onc.1204029. PMID 11175333.
- ↑ Chang, Nan-Shan (Mar. 2002). "The non-ankyrin C terminus of Ikappa Balpha physically interacts with p53 in vivo and dissociates in response to apoptotic stress, hypoxia, DNA damage, and transforming growth factor-beta 1-mediated growth suppression". J. Biol. Chem. 277 (12): 10323–31. doi:10.1074/jbc.M106607200. PMID 11799106.
- ↑ Seto, E; Usheva A, Zambetti G P, Momand J, Horikoshi N, Weinmann R, Levine A J, Shenk T (Dec. 1992). "Wild-type p53 binds to the TATA-binding protein and represses transcription". Proc. Natl. Acad. Sci. U.S.A. 89 (24): 12028–32. doi:10.1073/pnas.89.24.12028. PMID 1465435.
- ↑ Cvekl, A; Kashanchi F, Brady J N, Piatigorsky J (Jun. 1999). "Pax-6 interactions with TATA-box-binding protein and retinoblastoma protein". Invest. Ophthalmol. Vis. Sci. 40 (7): 1343–50. PMID 10359315.
- ↑ Wadhwa, Renu; Yaguchi Tomoko, Hasan Md K, Mitsui Youji, Reddel Roger R, Kaul Sunil C (Apr. 2002). "Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein". Exp. Cell Res. 274 (2): 246–53. doi:10.1006/excr.2002.5468. PMID 11900485.
- ↑ Badciong, James C; Haas Arthur L (Dec. 2002). "MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination". J. Biol. Chem. 277 (51): 49668–75. doi:10.1074/jbc.M208593200. PMID 12393902.
- ↑ Shvarts, A; Bazuine M, Dekker P, Ramos Y F, Steegenga W T, Merckx G, van Ham R C, van der Houven van Oordt W, van der Eb A J, Jochemsen A G (Jul. 1997). "Isolation and identification of the human homolog of a new p53-binding protein, Mdmx". Genomics 43 (1): 34–42. doi:10.1006/geno.1997.4775. PMID 9226370.
- ↑ Li, Muyang; Brooks Christopher L, Wu-Baer Foon, Chen Delin, Baer Richard, Gu Wei (Dec. 2003). "Mono- versus polyubiquitination: differential control of p53 fate by Mdm2". Science 302 (5652): 1972–5. doi:10.1126/science.1091362. PMID 14671306.
- ↑ Latonen, L; Taya Y, Laiho M (Oct. 2001). "UV-radiation induces dose-dependent regulation of p53 response and modulates p53-HDM2 interaction in human fibroblasts". Oncogene 20 (46): 6784–93. doi:10.1038/sj.onc.1204883. PMID 11709713.
- ↑ 159.0 159.1 Grossman, S R; Perez M, Kung A L, Joseph M, Mansur C, Xiao Z X, Kumar S, Howley P M, Livingston D M (Oct. 1998). "p300/MDM2 complexes participate in MDM2-mediated p53 degradation". Mol. Cell 2 (4): 405–15. doi:10.1016/S1097-2765(00)80140-9. PMID 9809062.
- ↑ Fang, S; Jensen J P, Ludwig R L, Vousden K H, Weissman A M (Mar. 2000). "Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53". J. Biol. Chem. 275 (12): 8945–51. doi:10.1074/jbc.275.12.8945. PMID 10722742.
- ↑ Liu, G; Schwartz J A, Brooks S C (Apr. 2000). "Estrogen receptor protects p53 from deactivation by human double minute-2". Cancer Res. 60 (7): 1810–4. PMID 10766163.
- ↑ Buschmann, T; Fuchs S Y, Lee C G, Pan Z Q, Ronai Z (Jun. 2000). "SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53". Cell 101 (7): 753–62. doi:10.1016/S0092-8674(00)80887-9. PMID 10892746.
- ↑ Chen, J; Marechal V, Levine A J (Jul. 1993). "Mapping of the p53 and mdm-2 interaction domains". Mol. Cell. Biol. 13 (7): 4107–14. PMID 7686617.
- ↑ Thut, C J; Goodrich J A, Tjian R (Aug. 1997). "Repression of p53-mediated transcription by MDM2: a dual mechanism". Genes Dev. 11 (15): 1974–86. PMID 9271120.
- ↑ Wawrzynow, Bartosz; Pettersson Susanne, Zylicz Alicja, Bramham Janice, Worrall Erin, Hupp Ted R, Ball Kathryn L (Apr. 2009). "A function for the RING finger domain in the allosteric control of MDM2 conformation and activity". J. Biol. Chem. 284 (17): 11517–30. doi:10.1074/jbc.M809294200. PMID 19188367.
- ↑ Ochocka, Anna Maria; Kampanis Petros, Nicol Samantha, Allende-Vega Nerea, Cox Miranda, Marcar Lynnette, Milne Diane, Fuller-Pace Frances, Meek David (Feb. 2009). "FKBP25, a novel regulator of the p53 pathway, induces the degradation of MDM2 and activation of p53". FEBS Lett. 583 (4): 621–6. doi:10.1016/j.febslet.2009.01.009. PMID 19166840.
- ↑ Chen, Deng; Zhang Jianbing, Li Mao, Rayburn Elizabeth R, Wang Hui, Zhang Ruiwen (Feb. 2009). "RYBP stabilizes p53 by modulating MDM2". EMBO Rep. 10 (2): 166–72. doi:10.1038/embor.2008.231. PMID 19098711.
- ↑ Hedström, Elisabeth; Issaeva Natalia, Enge Martin, Selivanova Galina (Feb. 2009). "Tumor-specific induction of apoptosis by a p53-reactivating compound". Exp. Cell Res. 315 (3): 451–61. doi:10.1016/j.yexcr.2008.11.009. PMID 19071110.
- ↑ Brenkman, Arjan B; de Keizer Peter L J, van den Broek Niels J F, Jochemsen A G, Burgering Boudewijn M Th (2008). "Mdm2 induces mono-ubiquitination of FOXO4". PLoS ONE 3 (7): e2819. doi:10.1371/journal.pone.0002819. PMID 18665269.
- ↑ Qiu, W; Wu J, Walsh E M, Zhang Y, Chen C-Y, Fujita J, Xiao Z-X J (Jul. 2008). "Retinoblastoma protein modulates gankyrin-MDM2 in regulation of p53 stability and chemosensitivity in cancer cells". Oncogene 27 (29): 4034–43. doi:10.1038/onc.2008.43. PMID 18332869.
- ↑ Muller, P; Hrstka R, Coomber D, Lane D P, Vojtesek B (May. 2008). "Chaperone-dependent stabilization and degradation of p53 mutants". Oncogene 27 (24): 3371–83. doi:10.1038/sj.onc.1211010. PMID 18223694.
- ↑ Watcharasit, Piyajit; Bijur Gautam N, Zmijewski Jaroslaw W, Song Ling, Zmijewska Anna, Chen Xinbin, Johnson Gail V W, Jope Richard S (Jun. 2002). "Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage". Proc. Natl. Acad. Sci. U.S.A. 99 (12): 7951–5. doi:10.1073/pnas.122062299. PMID 12048243.
- ↑ Young, Philip J; Day Patricia M, Zhou Jianhua, Androphy Elliot J, Morris Glenn E, Lorson Christian L (Jan. 2002). "A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy". J. Biol. Chem. 277 (4): 2852–9. doi:10.1074/jbc.M108769200. PMID 11704667.
- ↑ Wang, Chuangui; Chen Jiandong (Jan. 2003). "Phosphorylation and hsp90 binding mediate heat shock stabilization of p53". J. Biol. Chem. 278 (3): 2066–71. doi:10.1074/jbc.M206697200. PMID 12427754.
- ↑ Peng, Y; Chen L, Li C, Lu W, Chen J (Nov. 2001). "Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization". J. Biol. Chem. 276 (44): 40583–90. doi:10.1074/jbc.M102817200. PMID 11507088.
- ↑ McPherson, Lisa A; Loktev Alexander V, Weigel Ronald J (Nov. 2002). "Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53". J. Biol. Chem. 277 (47): 45028–33. doi:10.1074/jbc.M208924200. PMID 12226108.
- ↑ Huang, S M; Schönthal A H, Stallcup M R (Apr. 2001). "Enhancement of p53-dependent gene activation by the transcriptional coactivator Zac1". Oncogene 20 (17): 2134–43. doi:10.1038/sj.onc.1204298. PMID 11360197.
- ↑ Daniely, Yaron; Dimitrova Diana D, Borowiec James A (Aug. 2002). "Stress-dependent nucleolin mobilization mediated by p53-nucleolin complex formation". Mol. Cell. Biol. 22 (16): 6014–22. doi:10.1128/MCB.22.16.6014-6022.2002. PMID 12138209.
- ↑ Taniura, H; Matsumoto K, Yoshikawa K (Jun. 1999). "Physical and functional interactions of neuronal growth suppressor necdin with p53". J. Biol. Chem. 274 (23): 16242–8. doi:10.1074/jbc.274.23.16242. PMID 10347180.
- ↑ 180.0 180.1 Lee, Daeyoup; Kim Jin Woo, Seo Taegun, Hwang Sun Gwan, Choi Eui-Ju, Choe Joonho (Jun. 2002). "SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription". J. Biol. Chem. 277 (25): 22330–7. doi:10.1074/jbc.M111987200. PMID 11950834.
- ↑ An, Woojin; Kim Jaehoon, Roeder Robert G (Jun. 2004). "Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53". Cell 117 (6): 735–48. doi:10.1016/j.cell.2004.05.009. PMID 15186775.
- ↑ Pastorcic, M; Das H K (Nov. 2000). "Regulation of transcription of the human presenilin-1 gene by ets transcription factors and the p53 protooncogene". J. Biol. Chem. 275 (45): 34938–45. doi:10.1074/jbc.M005411200. PMID 10942770.
- ↑ Sørensen, T S; Girling R, Lee C W, Gannon J, Bandara L R, La Thangue N B (Oct. 1996). "Functional interaction between DP-1 and p53". Mol. Cell. Biol. 16 (10): 5888–95. PMID 8816502.
Additional images
 The DAXX Pathway |
External links
- "p53 Knowledgebase". Lane Group at the Institute of Molecular and Cell Biology (IMCB), Singapore. http://p53.bii.a-star.edu.sg/. Retrieved 2008-04-06.
- GeneReviews/NCBI/NIH/UW entry on Li-Fraumeni Syndrome
- TUMOR PROTEIN p53 @ OMIM
- P53 @ The Atlas of Genetics and Cytogenetics in Oncology and Haematology
- TP53 Gene @ GeneCards
- P53 News provided by insciences organisation
- David S. Goodsell (2002-07-01). "p53 Tumor Suppressor". Molecule of the Month. RCSB Protein Data Bank. http://www.pdb.org/pdb/static.do?p=education_discussion/molecule_of_the_month/pdb31_2.html. Retrieved 2008-04-06.
- Thierry Soussi. "p53 Web Site". http://p53.free.fr/. Retrieved 2008-04-06.
|
||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||